Nombre comercial: Somerix®
Denominación común internacional (DCI): Semaglutida
Forma farmacéutica, dosificación: Solución para administración subcutánea, 1.34 mg/mL (pluma jeringa 0.25 mg o 0.5 mg o 1 mg/dosis)
Grupo farmacoterapéutico: Medicamentos del sistema digestivo y metabolismo. Antidiabético. Medicamentos para reducir el nivel de glucosa en la sangre, excluyendo las insulinas. Análogo del péptido similar al glucagón tipo 1 (GLP-1) Semaglutida. Código ATC: A10BJ06
Indicaciones terapéuticas
Somerix® está indicado en el tratamiento de adultos con diabetes mellitus tipo 2, que no han sido controlados adecuadamente, como complemento de la dieta y el ejercicio
Información necesaria para el uso del medicamento
Contraindicaciones
- hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección “Composición del medicamento”
- cáncer medular de tiroides en el historial, incluido el familiar
- neoplasia endocrina múltiple (NEM) tipo 2
Precauciones de uso
No se recomienda el uso de semaglutida en pacientes con diabetes mellitus tipo 1 o para el tratamiento de la cetoacidosis diabética. Semaglutida no es un sustituto de la insulina.
No existe experiencia en pacientes con insuficiencia cardiaca crónica de clase IV (según la clasificación NYHA) y por lo tanto, el uso de semaglutida no se recomienda en estos pacientes.
Efectos gastrointestinales
El uso de agonistas del receptor de GLP-1 se puede asociar con reacciones adversas gastrointestinales. Esto se debe tener en consideración al tratar a pacientes con la función renal alterada, puesto que las náuseas, los vómitos y la diarrea pueden causar deshidratación que podría producir a su vez un deterioro de la función renal (ver sección “Reacciones adversas y su tratamiento”).
Pancreatitis aguda
Se ha observado pancreatitis aguda con el uso de agonistas del receptor de GLP-1. Se debe informar a los pacientes de los síntomas característicos de la pancreatitis aguda. Ante la sospecha de pancreatitis, se debe interrumpir el tratamiento con semaglutida. No se debe reanudar el tratamiento con semaglutida si se confirma pancreatitis. Se debe extremar la precaución en pacientes con antecedentes de pancreatitis.
Hipoglucemia
Los pacientes tratados con semaglutida en combinación con una sulfonilurea o insulina podrían presentar un riesgo mayor de hipoglucemia. Es posible disminuir el riesgo de hipoglucemia reduciendo la dosis de los medicamentos de sulfonilurea o de insulina al inicio del tratamiento con semaglutida (ver sección “Reacciones adversas y su tratamiento”).
Retinopatía diabética
En el caso de los pacientes con retinopatía diabética tratados con insulina y semaglutida, se ha observado un riesgo mayor de desarrollar la retinopatía diabética (ver sección “Reacciones adversas y su tratamiento”). Se debe extremar la precaución al usar semaglutida en pacientes con retinopatía diabética en tratamiento con insulina. Es preciso monitorizar estrechamente a estos pacientes, así como tratarlos según las directrices clínicas correspondientes. La mejora rápida del control glucémico se ha asociado con un empeoramiento temporal de la retinopatía diabética, pero no se pueden excluir otros mecanismos.
Colelitiasis y colecistitis
Se han notificado enfermedades agudas de la vesícula biliar, como colelitiasis (cálculos en la vesícula biliar) o colecistitis, en estudios con agonistas de los receptores del GLP-1, tanto en el periodo posterior a la comercialización como en estudios controlados con placebo. Ante la sospecha de litiasis biliar, se debe realizar ensayos de la vesícula biliar y la observación clínica correspondiente. Es preciso monitorizar estrechamente a estos pacientes, así como tratarlos según las directrices clínicas correspondientes.
Riesgo de aspiración o neumonía por aspiración
Al realizar las intervenciones quirúrgicas bajo anestesia general o sedación profunda en los pacientes que toman los agonistas del GLP-1, existe el riesgo de aspiración pulmonar o neumonía por aspiración debido al vaciamiento gástrico retardado y volumen gástrico residual. Los datos disponibles son insuficientes para hacer recomendaciones sobre la reducción del riesgo de aspiración pulmonar durante la anestesia general o sedación profunda en pacientes que toman semaglutida. Se desconoce si modificar el período de ayuno preoperatorio o suspender temporalmente el uso de semaglutida podría reducir la frecuencia de vaciamiento gástrico retardado. Si los pacientes están tomando semaglutida, deben informar al personal médico ANTES de cualquier intervención quirúrgica o procedimiento con sedación.
Interacción con otros medicamentos
Semaglutida retrasa el vaciamiento gástrico y puede afectar a la tasa de absorción de medicamentos orales administrados de forma concomitante. Semaglutida se debe utilizar con precaución en pacientes en tratamiento con medicamentos orales que requieren una absorción gastrointestinal rápida.
Paracetamol
Semaglutida retrasa la velocidad de vaciamiento gástrico tal como determinó la farmacocinética de paracetamol durante una prueba de comida estándar. El AUC0–60 min y la Cmax de paracetamol se redujeron en un 27 % y un 23 %, respectivamente tras el uso concomitante de 1 mg de semaglutida. La exposición total de paracetamol (AUC0–5 h) no se vio afectada. No es necesario un ajuste de dosis de paracetamol cuando se administra con semaglutida.
Anticonceptivos orales
Semaglutida no disminuye el efecto de los anticonceptivos orales, ya que semaglutida no modificó de una forma clínicamente significativa la exposición general de etinilestradiol ni de levonorgestrel tras la administración conjunta de un medicamento anticonceptivo oral combinado (0.03 mg de etinilestradiol/0.15 mg de levonorgestrel) y semaglutida. La exposición de etinilestradiol no se vio afectada; se observó un aumento del 20 % en la exposición de levonorgestrel en concentración de equilibrio. La Cmaxmax no se vio afectada por ninguno de los compuestos.
Atorvastatina
Semaglutida no modificó la exposición general de atorvastatina tras la administración de una dosis única de atorvastatina (40 mg). La Cmax de atorvastatina se redujo en un 38 %. Se concluyó que esta reducción no era clínicamente significativa.
Digoxina
Semaglutida no modificó la exposición general ni la Cmax de digoxina tras la administración de una dosis única de digoxina (0.5 mg).
Metformina
Semaglutida no modificó la exposición general ni la Cmax de metformina tras la administración de 500 mg de metformina dos veces al día durante 3.5 días.
Warfarina
Semaglutida no modificó la exposición general ni la Cmax de R- y S-warfarina tras la administración de una dosis única de warfarina (25 mg), asimismo, el efecto farmacodinámico de warfarina, determinado por el índice internacional normalizado (INR), no se vio afectado de una forma clínicamente significativa. No obstante, al inicio del tratamiento con semaglutida en pacientes tratados con warfarina u otros derivados de la cumarina, se recomienda un control frecuente de INR.
Si utiliza insulina, su médico le explicará cómo reducir la dosis de insulina y le recomendará controlar sus niveles de glucosa en sangre con más frecuencia para evitar la hiperglucemia (niveles altos de glucosa en sangre) o la cetoacidosis diabética (una complicación de la diabetes mellitus que ocurre cuando el cuerpo no tiene suficiente insulina para descomponer la glucosa).
Advertencias especiales
Pacientes de edad avanzada
No es necesario un ajuste de dosis en función de la edad. La experiencia terapéutica en pacientes ≥75 años es limitada.
Pacientes con insuficiencia renal
No es necesario un ajuste de dosis en pacientes con insuficiencia renal leve, moderada o grave.
La experiencia con el uso de semaglutida en pacientes con insuficiencia renal grave es limitada.
No se recomienda el uso de semaglutida en pacientes con insuficiencia renal en etapa terminal.
Pacientes con insuficiencia hepática
No es necesario un ajuste de dosis en pacientes con insuficiencia hepática. La experiencia con el uso de semaglutida en pacientes con insuficiencia hepática grave es limitada. Se debe extremar la precaución al tratar a estos pacientes con semaglutida.
Contenido de sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, por lo tanto puede considerarse “exento de sodio”.
Población pediátrica
No se recomienda el uso del medicamento en niños y adolescentes menores de 18 años, ya que no se ha establecido todavía la seguridad y eficacia del medicamento en este grupo de edad.
Embarazo y lactancia
Si está embarazada o amamantando, sospecha que está embarazada o planea quedar embarazada, consulte a su médico antes de usar este medicamento.
No se debe utilizar el medicamento durante el embarazo, ya que no se ha estudiado el efecto en el feto. Por lo tanto, se recomienda utilizar métodos anticonceptivos durante el tratamiento con Somerix®. El tratamiento con el medicamento se debe interrumpir al menos 2 meses antes de un embarazo planeado. Si queda embarazada mientras toma el medicamento, consulte con su médico, ya que puede ser necesario cambiar el tratamiento.
El medicamento no se debe utilizar durante la lactancia, ya que no se sabe si se excreta en la leche materna.
Efectos sobre la capacidad para conducir y utilizar máquinas potencialmente peligrosos
Cuando se utilice en combinación con una sulfonilurea o una insulina, se recomienda tomar las precauciones para evitar niveles bajos de glucosa en la sangre (hipoglucemia), ya que la capacidad de concentración puede disminuir. No puede conducir y utilizar máquinas, si tiene algún signo de nivel bajo de glucosa en sangre. Consulte las secciones ”Información necesaria para el uso del medicamento” y “Reacciones adversas y su tratamiento” para conocer los signos de un nivel bajo de glucosa en la sangre. Consulte a su médico para obtener más información.
Recomendaciones de uso
Siempre use el medicamento de acuerdo con las recomendaciones de su médico. Consulte a su médico si tiene dudas.
Posología
No cambie su dosis sin la recomendación de su médico.
Forma de administración
Somerix® es un solución para administración subcutánea. No se debe administrar por vía intravenosa ni intramuscular.
La información detallada se describe en la sección “Instrucciones de uso médico del medicamento Somerix®”.
Frecuencia de uso y la hora de administración
Se recomienda que anote en la caja el día de la semana elegido (por ejemplo, miércoles) y escriba la fecha de cada inyección, para ayudarle a recordar que se inyecte este medicamento solo una vez a la semana.
El día de administración semanal del medicamento puede cambiarse si es necesario, siempre que el tiempo entre dos dosis sea de al menos 3 días. Después de seleccionar un nuevo día de administración, se debe continuar con la administración de una vez a la semana.
Medidas a tomar en caso de sobredosis
Si ha tomado una dosis de Somerix® más de lo que debería, consulte inmediatamente a su médico. Puede experimentar reacciones adversas como náuseas.
Medidas a tomar si se olvida de una o más dosis del medicamento
En caso de que se olvide de inyectar la dosis:
No se debe administrar una dosis doble de Somerix® para compensar la dosis omitida.
Síntomas de abstinencia
No deje de tomar Somerix® sin consultar primero con su médico. Si deja de tomar el medicamento, su glucosa en sangre puede aumentar.
Recomendaciones para consultar a un profesional sanitario sobre el modo de administración del medicamento
Si tiene algunas preguntas sobre el uso de Somerix®, consulte a un profesional sanitario sobre el modo de administración del medicamento.
Reacciones adversas y su tratamiento
Las frecuencias de reacciones adversas se definen del siguiente modo: muy frecuentes (≥1/10); frecuentes (≥1/100 a <1 /10); poco frecuentes (≥1/1 000 a <1 /100); raras (≥1/10 000 a <1 /1 000); muy raras (<1 /10 000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Muy frecuentes
- Hipoglucemiaa cuando se utiliza con insulina o sulfonilurea
- Náuseas
- Diarrea
Frecuentes
- Hipoglucemiaа cuando se utiliza con otros antidiabéticos orales (ADO)
- Apetito disminuido
- Mareo
- Complicaciones de la retinopatía diabéticab
- Vómitos
- Dolor abdominal
- Distensión abdominal
- Estreñimiento
- Dispepsia
- Gastritis
- Enfermedad por reflujo gastroesofágico
- Eructos
- Flatulencia
- Colelitiasis
- Fatiga
- Aumento de concentración de lipasa
- Aumento de concentración de amilasa
- Peso disminuido
Poco frecuentes
- Disgeusia
- Aumento de la frecuencia cardiaca
- Inflamación del páncreas (pancreatitis aguda)
- Reacciones en el lugar de inyección
- Reacciones alérgicas como erupción cutánea, picazón o urticaria
Poco frecuentes
- Reacción anafiláctica.
Frecuencia no conocida
- Angioedema
Busque atención médica o informe al médico de inmediato, si experimenta cualquiera de los síntomas siguientes: problemas respiratorios, hinchazón de la cara, labios, lengua y/o garganta con dificultad para tragar y palpitaciones.
⁽ᵃ⁾La hipoglucemia se define como grave (precisa la ayuda de otra persona) o sintomática en combinación con un nivel de glucosa en sangre <3.1 mmol/L.
⁽ᵇ⁾Las complicaciones de la retinopatía diabética incluyen: necesidad de fotocoagulación retiniana, tratamiento con agentes intravítreos, hemorragia del vítreo y ceguera relacionada con la diabetes (poco frecuentes). Frecuencia basada en el ensayo de resultados cardiovasculares.
Descripción de las reacciones adversas seleccionadas
Hipoglucemia
No se observaron episodios de hipoglucemia grave cuando semaglutida se usó en monoterapia. La hipoglucemia grave se observó principalmente cuando semaglutida se usó con una sulfonilurea (1.2 % de los sujetos; 0.03 episodios/paciente-año) o insulina (1.5 % de los sujetos; 0.02 episodios/paciente-año). Se observaron pocos episodios (0.1 % de los sujetos; 0.001 episodios/paciente-año) al administrar semaglutida en combinación con medicamentos hipoglucemiantes orales distintos de las sulfonilureas.
Reacciones adversas gastrointestinales
En los pacientes tratados con 0.5 mg y 1 mg de semaglutida, se produjeron náuseas en un 17.0 % y en un 19.9 %, respectivamente, diarrea en un 12.2 % y en un 13.3 % y vómitos en un 6.4 % y en un 8.4 %. La mayoría de los episodios fueron de leves a moderados en gravedad y de corta duración. Los episodios causaron la interrupción del tratamiento en un 3.9 % y un 5 % de los pacientes. Los episodios se notificaron con mayor frecuencia durante los primeros meses del tratamiento.
Los pacientes con un peso corporal bajo pueden experimentar más acontecimientos adversos gastrointestinales al ser tratados con semaglutida.
Complicaciones de la retinopatía diabética
En un estudio clínico de 2 años de duración participaron 3297 pacientes con diabetes mellitus tipo 2 de larga duración, alto riesgo cardiovascular y nivel de glucosa en sangre no controlado adecuadamente. En este estudio, las complicaciones evaluadas de la retinopatía diabética ocurrieron en más pacientes tratados con semaglutida (3.0 %) que en los que recibieron placebo (1.8 %). Esto se observó en pacientes tratados con insulina, que tenían antecedentes de retinopatía diabética. La diferencia de tratamiento apareció de manera temprana y persistió a lo largo del estudio. La evaluación sistemática de la complicación de la retinopatía diabética solo se realizó en el estudio de resultados cardiovasculares. En los estudios clínicos de hasta 1 año de duración en los que participaron 4807 pacientes con diabetes mellitus tipo 2, se notificaron acontecimientos adversos relacionados con retinopatía diabética en proporciones similares en pacientes tratados con semaglutida (1.7 %) y con los comparadores (2.0 %).
Interrupción prematura a causa de un acontecimiento adverso
La incidencia de interrupción del tratamiento a causa de acontecimientos adversos fue del 6.1 % y del 8.7 % entre los pacientes tratados con 0.5 mg y 1 mg de semaglutida, respectivamente, frente al 1.5 % con placebo. Los acontecimientos adversos más frecuentes que causaron la interrupción del tratamiento fueron gastrointestinales.
Reacciones en el lugar de inyección
Se han notificado reacciones en el lugar de inyección (por ejemplo, erupción o eritema en el área de inyección) en el 0.6 % y el 0.5 % de los pacientes tratados con 0.5 mg y 1 mg de semaglutida, respectivamente. Estas reacciones, por lo general, han sido leves.
Inmunogenicidad
De acuerdo a las propiedades potencialmente inmunogénicas de los medicamentos que contienen proteínas o péptidos, los pacientes pueden desarrollar anticuerpos tras el tratamiento con semaglutida. La proporción de pacientes con un resultado positivo en el análisis de anticuerpos antisemaglutida en cualquier punto temporal posterior al inicio del estudio fue baja (1–2 %) y, al final del estudio, ningún paciente presentó anticuerpos neutralizantes antisemaglutida ni anticuerpos antisemaglutida con efecto neutralizante de GLP-1 endógeno.
Aumento de la frecuencia cardiaca
Un aumento de la frecuencia cardiaca se observa con los agonistas del receptor de GLP-1. En los estudios de fase 3a se observaron, en los pacientes tratados con semaglutida, aumentos de 1 a 6 latidos por minuto (lpm) de media, partiendo de frecuencias basales de 72 a 76 lpm. En los estudios a largo plazo en pacientes con alto riesgo cardiovascular, el 16 % de los pacientes tratados con semaglutida tuvieron un aumento en la frecuencia cardiaca de más de 10 lpm comparado con el 11 % de los pacientes tratados con placebo, tras dos años de tratamiento.
Si tiene reacciones adversas al medicamento, notifique a un profesional sanitario, profesional farmacéutico, al Sistema Nacional de Farmacovigilancia o al Departamento de farmacovigilancia del Titular de Registro Sanitario. Al informar sobre las reacciones adversas, Usted ayuda a obtener más información sobre la seguridad del medicamento.
Propiedades farmacológicas
Propiedades farmacodinámicas
Grupo farmacoterapéutico: medicamentos utilizados en la diabetes. Medicamentos para reducir el nivel de glucosa en la sangre, excluyendo las insulinas. Análogo del péptido similar al glucagón tipo 1 (GLP-1) Semaglutida.
Código ATC: A10BJ06
Mecanismo de acción
Semaglutida es un análogo de GLP-1 con un 94 % de homología de secuencia con GLP-1 humano. Semaglutida actúa como un agonista del receptor de GLP-1 que se une de forma selectiva al receptor de GLP-1 (el objetivo de GLP-1 nativo) y lo activa.
GLP-1 es una hormona fisiológica que desempeña diversas funciones en la regulación de la glucosa y del apetito, así como en el sistema cardiovascular. Los efectos sobre la concentración de glucosa y el apetito están mediados específicamente por los receptores de GLP-1 presentes en el páncreas y el cerebro.
Semaglutida reduce la glucosa en sangre de un modo dependiente de la glucosa, mediante la estimulación de la secreción de insulina y la disminución de la secreción de glucagón cuando la glucosa en sangre es elevada. El mecanismo de disminución de la glucosa en sangre también implica un ligero retraso en el vaciamiento gástrico en la fase posprandial temprana. Durante la hipoglucemia, semaglutida disminuye la secreción de insulina y no afecta a la secreción de glucagón.
Semaglutida reduce el peso corporal y la masa grasa corporal mediante la reducción de la ingesta calórica, incluso una reducción general del apetito. Además, semaglutida reduce la preferencia por alimentos ricos en grasas.
Los receptores de GLP-1 también se expresan en el corazón, el sistema vascular, el sistema inmunitario y los riñones. En los estudios clínicos realizados, semaglutida ejerció un efecto beneficioso en la concentración de los lípidos plasmáticos, disminuyó la presión arterial sistólica y redujo la inflamación. En los ensayos realizados en animales, semaglutida atenuó el desarrollo de ateroesclerosis mediante la prevención de la progresión de la placa aórtica y la reducción de la inflamación en la placa.
Efectos farmacodinámicos
Todas las evaluaciones farmacodinámicas se realizaron transcurridas 12 semanas de tratamiento (incluido el escalado de la dosis) en estado estacionario con 1 mg de semaglutida una vez a la semana.
Glucosa en ayunas y posprandial
Semaglutida reduce las concentraciones de glucosa en ayunas y posprandial. En pacientes con diabetes tipo 2, el tratamiento con semaglutida 1 mg logró reducciones de la glucosa en términos de cambio absoluto con respecto al valor inicial (mmol/L) y de reducción relativa, en comparación con el placebo (%) para la glucosa en ayunas (1.6 mmol/L; reducción del 22 %), glucosa posprandial a las 2 horas (4.1 mmol/L; reducción del 37 %), concentración media de glucosa a las 24 horas (1.7 mmol/L; reducción del 22 %) y fluctuación de la glucosa posprandial durante 3 comidas (0.6–1.1 mmol/L). Semaglutida redujo la glucosa en ayunas después de la administración de la primera dosis.
Función de las células beta y secreción de insulina
Semaglutida mejora la función de las células beta. En comparación con placebo, semaglutida mejoró la respuesta a la insulina durante la primera y la segunda fase con un aumento que triplicó y duplicó ésta, respectivamente, y aumentó la capacidad secretora máxima de las células beta en pacientes con diabetes tipo 2. Además, el tratamiento con semaglutida aumentó las concentraciones de insulina en ayunas en comparación con placebo.
Secreción de glucagón
Semaglutida disminuye las concentraciones de glucagón en ayunas y posprandial. En pacientes con diabetes tipo 2, en comparación con placebo, semaglutida logró las siguientes reducciones relativas de glucagón: glucagón en ayunas (8–21 %), respuesta de glucagón posprandial (14–15 %) y concentración media de glucagón a las 24 horas (12 %).
Secreción de glucagón e insulina dependiente de la glucosa
Semaglutida disminuyó las concentraciones elevadas de glucosa en sangre mediante la estimulación de la secreción de insulina y la disminución de la secreción de glucagón de un modo dependiente de la glucosa. Con semaglutida, la tasa de secreción de insulina en pacientes con diabetes tipo 2 fue comparable a la de los sujetos sanos.
Durante la hipoglucemia inducida, en comparación con placebo, semaglutida no alteró las respuestas contrarreguladoras del aumento de glucagón y tampoco afectó a la disminución de péptido C en pacientes con diabetes tipo 2.
Vaciamiento gástrico
Semaglutida causó un ligero retraso del vaciamiento gástrico en la fase posprandial temprana, reduciendo así la velocidad a la que la glucosa aparece en la circulación después de las comidas.
Apetito, ingesta calórica y elecciones alimentarias
En comparación con placebo, semaglutida redujo la ingesta calórica de 3 comidas consecutivas (a voluntad) en un 18–35 %. Esto se vio favorecido por una supresión del apetito inducida por semaglutida en estado de ayunas así como en el posprandial, un mejor control de la ingesta, menor deseo de ciertos tipos de alimentos y una preferencia relativamente menor por alimentos ricos en grasas.
Lípidos en ayunas y posprandiales
En comparación con placebo, semaglutida redujo las concentraciones de triglicéridos y colesterol de lipoproteínas de muy baja densidad (VLDL) en ayunas en un 12 % y un 21 %, respectivamente. Los triglicéridos y colesterol VLDL posprandiales en respuesta a una comida rica en grasas se redujeron en más de 40 %.
Electrofisiología cardíaca (QTc)
El efecto de semaglutida en la repolarización cardiaca se evaluó en un ensayo de monitorización del intervalo QTc. Semaglutida no prolongó los intervalos QTc en niveles de dosis supraterapéuticos (hasta 1.5 mg en estado estacionario).
Propiedades farmacocinéticas
En comparación con GLP-1 nativo, semaglutida tiene una semivida prolongada de aproximadamente 1 semana, por lo que es idónea para la administración subcutánea una vez a la semana. El mecanismo principal de prolongación es la unión a albúmina, que propicia una disminución del aclaramiento renal y protege de la degradación metabólica. Asimismo, semaglutida es resistente frente a la degradación por la enzima dipeptidil peptidasa tipo IV (DPP-4).
Absorción
La concentración máxima se alcanzó entre 1 y 3 días después de la dosis. La exposición en estado estacionario se alcanzó después de 4–5 semanas de la administración una vez a la semana. En pacientes con diabetes tipo 2, las concentraciones medias en estado estacionario tras la administración subcutánea de 0.5 mg y 1 mg de semaglutida fueron de aproximadamente 16 nmol/L y 30 nmol/L, respectivamente. Para las dosis de 0.5 mg y 1 mg, la exposición a semaglutida aumentó de forma proporcional a la dosis. Asimismo, se logró una exposición similar con la administración de semaglutida subcutánea en el abdomen, el muslo y el brazo. La biodisponibilidad absoluta de semaglutida después de la administración subcutánea fue del 89 %.
Distribución
El volumen medio de distribución de semaglutida tras su administración subcutánea en pacientes con diabetes tipo 2 fue de aproximadamente 12.5 L. Semaglutida se encontraba ampliamente unida a albúmina en plasma (>99 %).
Biotransformación
Antes de su excreción, semaglutida se metaboliza en gran medida mediante proteólisis del esqueleto peptídico y beta‑oxidación secuencial de la cadena lateral del ácido graso. Se cree que la enzima denominada endopeptidasa neutra interviene en el metabolismo de semaglutida.
Eliminación
En un ensayo que empleó una única dosis subcutánea de semaglutida radiomarcada, se determinó que las principales vías de excreción de los productos relacionados con semaglutida eran la orina y las heces. Aproximadamente 2/3 de estos productos se excretaron en orina y aproximadamente 1/3 en heces. Alrededor del 3 % de la dosis de semaglutida se excretó en forma intacta en orina. En pacientes con diabetes mellitus tipo 2, el aclaramiento de semaglutida fue de 0.05 L/h aproximadamente. Con una semivida de eliminación aproximada de 1 semana, semaglutida permanecerá en la circulación durante un tiempo aproximado de 5 semanas después de la última dosis.
Farmacocinética poblacional
Edad avanzada
La edad no tuvo ningún efecto sobre la farmacocinética de semaglutida según los resultados de los ensayos de fase 3a realizados que incluyeron a pacientes de 20 a 86 años de edad.
Sexo, raza y etnia
El sexo, la raza (blanca, negra o afroamericana, asiática) y la etnia (hispana o latina, no hispana o latina) no tuvieron ningún efecto sobre la farmacocinética de semaglutida.
Peso corporal
El peso corporal tiene efecto en la exposición de semaglutida. Cuanto mayor es el peso corporal, menor es la exposición; una diferencia del 20 % en el peso corporal entre individuos se traducirá en una diferencia aproximada del 16 % en la exposición. Las dosis de 0.5 mg y 1 mg de semaglutida proporcionan una exposición sistémica adecuada en el rango de peso corporal de 40–198 kg.
Insuficiencia renal
La insuficiencia renal no tuvo ningún efecto clínicamente significativo sobre la farmacocinética de semaglutida. Esto se constató comparando los efectos de una dosis única de 0.5 mg de semaglutida en pacientes con diversos grados de insuficiencia renal (leve, moderada, grave o pacientes en diálisis) con sujetos con función renal normal. Los datos de los estudios de fase 3a confirmaron esto mismo en sujetos con diabetes tipo 2 e insuficiencia renal. Sin embargo, la experiencia en pacientes con enfermedad renal en etapa terminal fue limitada.
Insuficiencia hepática
La insuficiencia hepática no tuvo ningún efecto en la exposición de semaglutida. La farmacocinética de semaglutida se evaluó en pacientes con diversos grados de insuficiencia hepática (leve, moderada y grave) en comparación con sujetos con función hepática normal en un ensayo de dosis única de 0.5 mg de semaglutida.
Niños
Semaglutida no se ha estudiado en pacientes pediátricos.
Información adicional
Composición del medicamento
1 mL del medicamento contiene principio activo: semaglutida*, 1.34 mg,
*análogo humano del péptido similar al glucagón tipo 1 (GLP-1) producido por síntesis química.
Una pluma jeringa precargada contiene 4 mg de semaglutida en 3 mL de solución. Cada dosis contiene 0.25 mg de semaglutida en 0.19 mL de solución o 0.5 mg de semaglutida en 0.37 mL de solución o 1 mg de semaglutida en 0.74 mL de solución.
excipientes:
hidrofosfato disódico dihidratado, propilenglicol, fenol, ácido clorhídrico diluido 10% y/o solución de hidróxido de sodio 10% (para ajuste de pH), agua para inyección
Aspecto, olor y sabor
Solución incolora o casi incolora.
Presentación y envase
3 mL del medicamento en cartuchos de vidrio neutro incoloro con émbolo de goma y un capuchón de aluminio con una hoja de caucho.
El cartucho se coloca en una pluma jeringa desechable multidosis de plástico para inyección múltiple. Al cuerpo de cada pluma jeringa se le pega una etiqueta de película de polipropileno.
1 pluma jeringa desechable multidosis precargada para inyección múltiple y 1 caja de cartón que contiene 4 agujas desechables colocadas en un soporte de cartón para agujas, con instrucciones de uso médico del medicamento, instrucciones de uso de la pluma jeringa y una tarjeta de inserción para el control de la administración del medicamento, se colocan en una caja de cartón.
Periodo de almacenamiento
2 años.
Período de vencimiento tras la primera apertura: 6 semanas.
¡No usar después de la fecha de vencimiento!
La fecha de caducidad es el último día de un mes indicado.
Condiciones de conservación
Conservar entre 2 °C y 8 °C (en el refrigerador), pero no cerca del componente del congelador. Proteger de la luz. No congelar.
Tras la primera apertura: conservar por debajo de 30 °C o en el refrigerador (2 8 °C). No congelar. Conservar la pluma jeringa con el capuchón puesto para protegerla de la luz.
No conservar la pluma jeringa con la aguja adjunta.
¡Mantener fuera del alcance de los niños!
Condiciones de dispensación
Se dispensa bajo receta médica
Información del fabricante
“GEROPHARM”, S.L.
UnoPen GM-B1 es una pluma jeringa desechable precargada para inyección múltiple, que contiene un agente hipoglucémico, un análogo del péptido similar al glucagón tipo 1 (GLP-1), semaglutida. La pluma jeringa le permite administrar dosis de 0.25 mg, 0.5 mg y 1 mg. La pluma jeringa está diseñada para titulación de la dosis de 0.25 mg y mantener una dosis terapéutica de 0.5 mg y 1 mg. Una pluma jeringa contiene 3 mL de solución.
Opciones de uso de la pluma jeringa:
Opción 1 - titulación de dosis
4 inyecciónes de 0.25 mg
4 inyecciónes de 0.5 mg
1 inyección de 1.0 mg
Opción 2 - mantenimiento de la dosis terapéutica
4 inyecciónes de 1.0 mg
La pluma jeringa está diseñada para ser utilizada con agujas desechables WellFine, Dexfine y Verifine. El envase de Somerix® incluye 4 agujas WellFine 4 mm 32G (Dexfine 4 mm 32G o Verifine 4 mm 32G).
Este medicamento se puede administrar con agujas de hasta 8 mm de longitud.
Utilice siempre una aguja nueva para cada inyección. ¡Después de la inyección, la pluma jeringa debe almacenarse y transportarse sin la aguja! Así se puede evitar que las agujas se atasquen, la contaminación, las infecciones, la pérdida de solución y las dosificaciones inexactas.
Las agujas deben desecharse según las instrucciones de autoridades locales y las normas de uso de materiales potencialmente infectados.
La pluma jeringa está destinada para ser utilizada por una sola persona. La pluma jeringa no debe ser entregada a terceros.
Nunca utilice la pluma jeringa Somerix® si la solución no tiene un aspecto transparente e incoloro.
No exponga las plumas jeringa a temperaturas bajas (inferiores a +2 °C) ni altas (superiores a +30 °C). No coloque las plumas jeringa en el congelador. ¡Las plumas jeringa no deben congelarse!
Una vez la pluma jeringa está vacía, se debe desechar (no intente usar la pluma una vez más y rellenarla).
Lo mejor es transportar las plumas jeringa a temperaturas altas/bajas en un estuche térmico/bolsa especial (por ejemplo, el estuche térmico original de “GEROPHARM”, S.L.).
Mantenga siempre la pluma jeringa y las agujas fuera de la vista y del alcance de otras personas, especialmente de los niños
No trate de reparar la pluma. En caso de rotura, notifique a la organización que se encarga de recibir las quejas de los consumidores, indicada en las instrucciones de uso médico del medicamento.
Antes de la primera inyección, lea atentamente las instrucciones de uso de las plumas desechables precargadas UnoPen GM-B1.
Consulte a su médico (profesional) sobre el uso de la pluma precargada. Pídale al médico que demuestre el uso correcto de la pluma jeringa. La primera inyección de este medicamento deberá administrarse bajo la supervisión de un médico o enfermera.
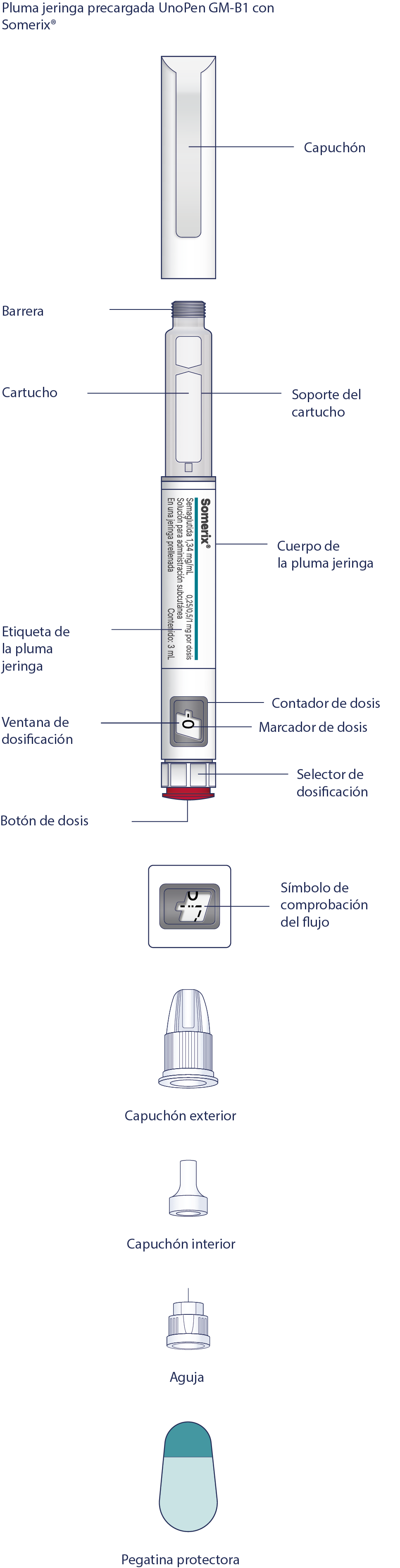
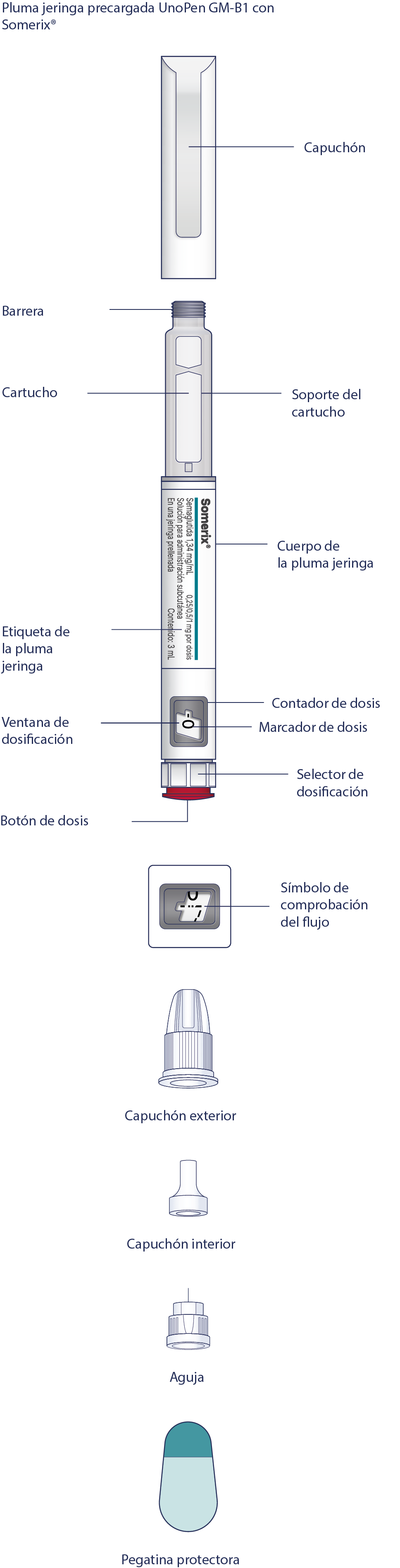
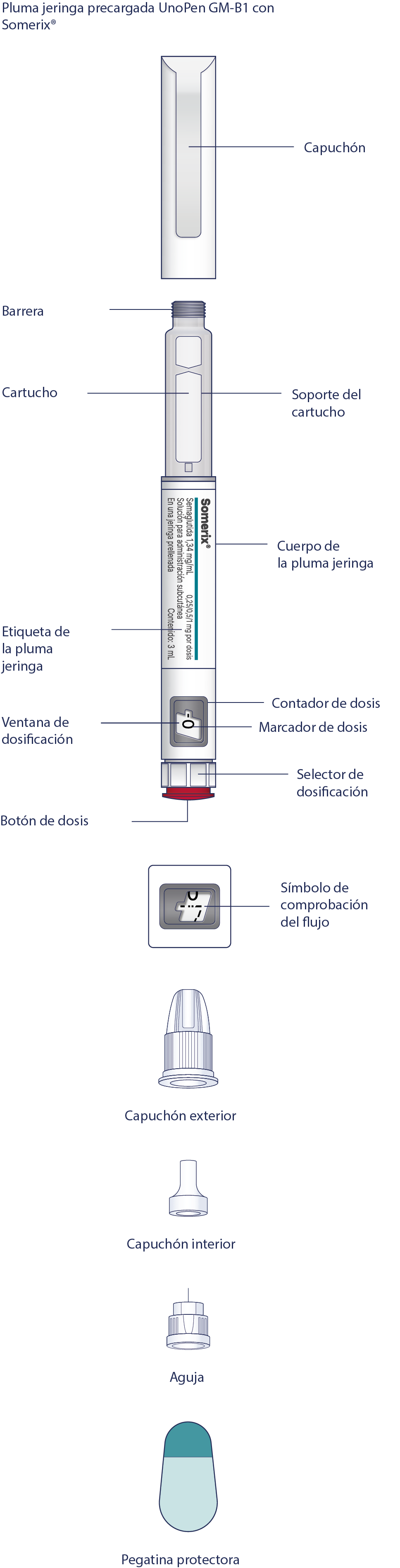
Lea atentamente la etiqueta de la pluma jeringa y asegúrese de usar el medicamento que le recetó el médico en la dosis necesaria, también compruebe la fecha de caducidad del medicamento. Preste atención a las siguientes ilustraciones para conocer las características y los componentes de la pluma jeringa.
Antes de empezar a utilizar su pluma jeringa, compruebe si hay algunos daños mecánicos visibles y pérdidas (indicadores de que el sello del cartucho está dañado). Si no está seguro de que la pluma jeringa esté en buen estado o no esté dañada, nunca la use. Compruebe siempre la pluma jeringa antes de cada inyección.
Siga atentamente las instrucciones de uso de la pluma jeringa: evite la caída de la pluma y la influencia de factores externos (exposición térmica, luz solar directa, daños mecánicos, etc.). De producirse un daño, es necesario comenzar a utilizar una pluma jeringa nueva.
Si es usted invidente o tiene visión reducida y no puede leer el contador de dosis de la pluma, debe usar una pluma jeringa bajo la supervisión de personal médico, parientes o una persona sin problemas de visión que están formados en el uso de la pluma jeringa precargada.
Las personas que atienden a los pacientes deben tener mucho cuidado cuando manejen agujas usadas para evitar pinchazos accidentales e infecciones.
La pluma jeringa precargada UnoPen GM-B1 con Somerix®
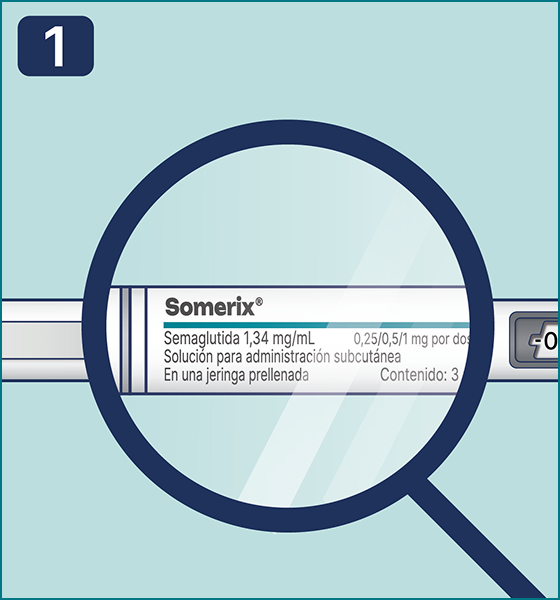
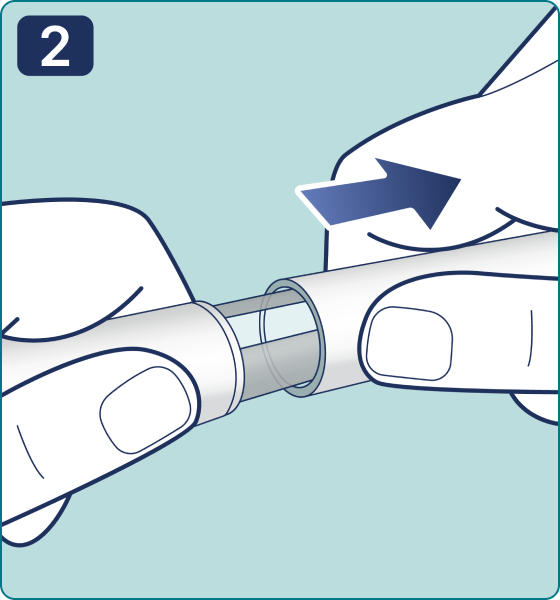
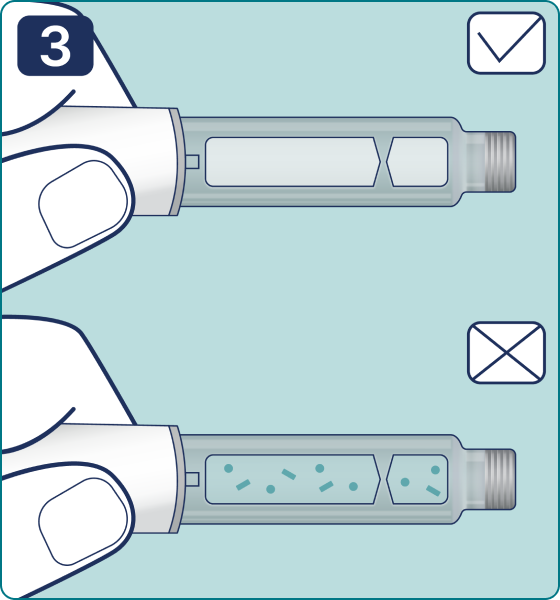
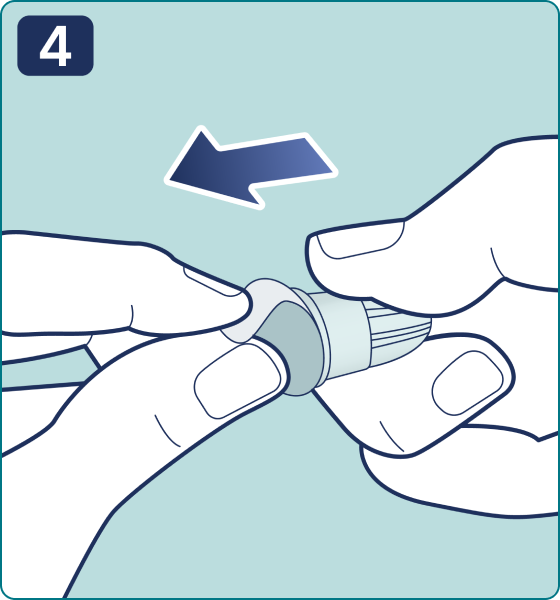
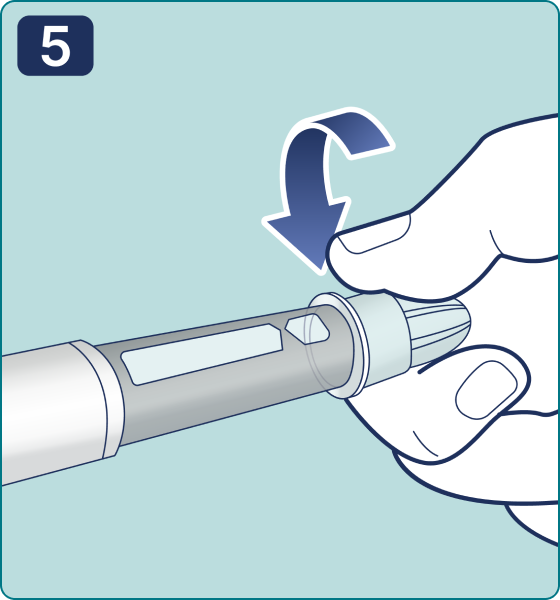
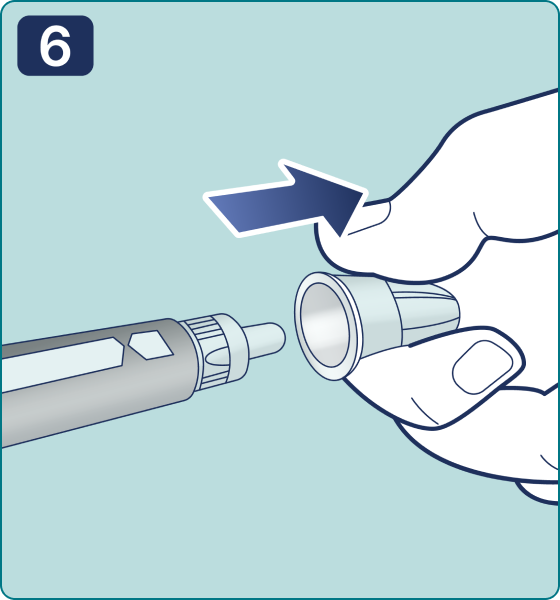
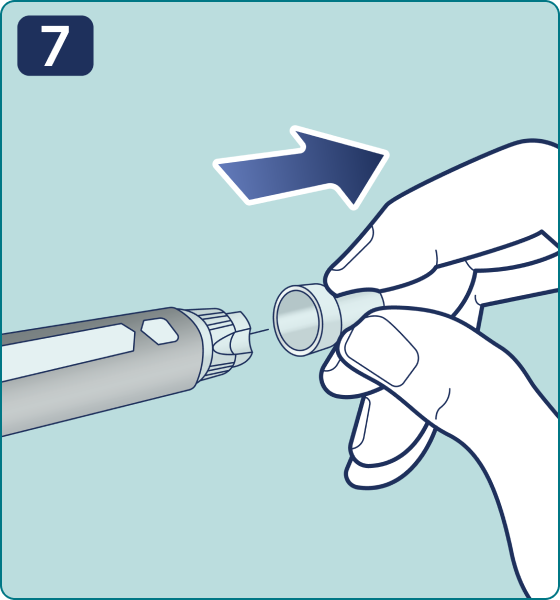
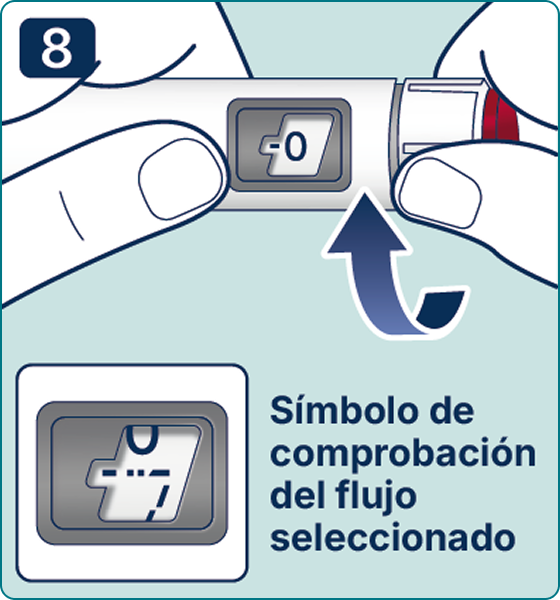
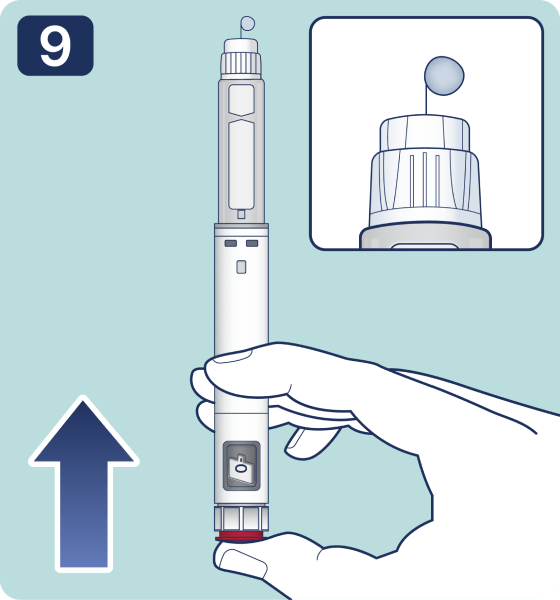
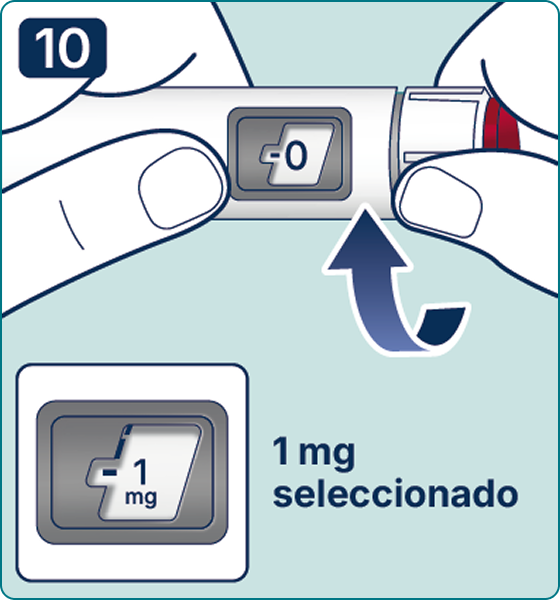
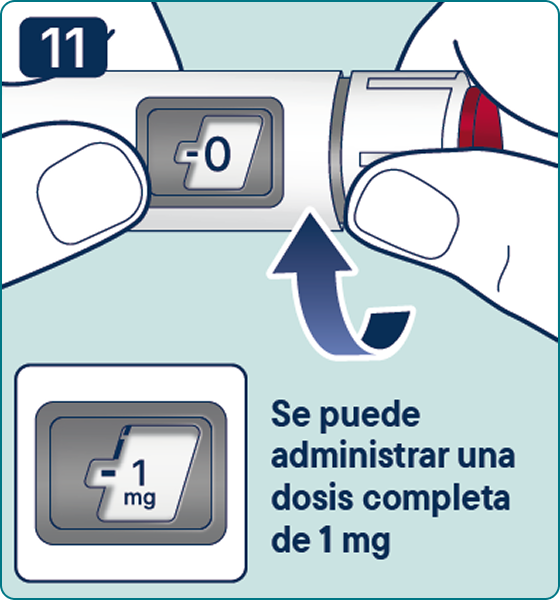
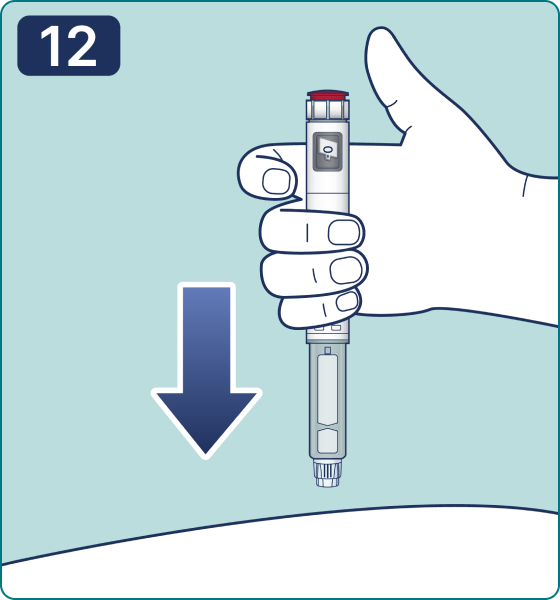
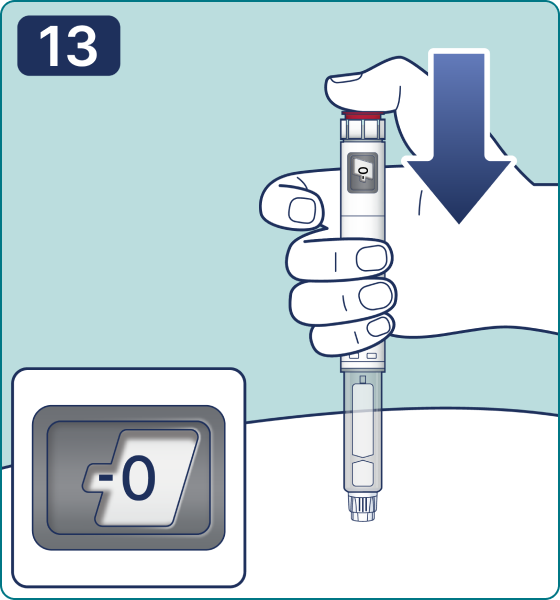
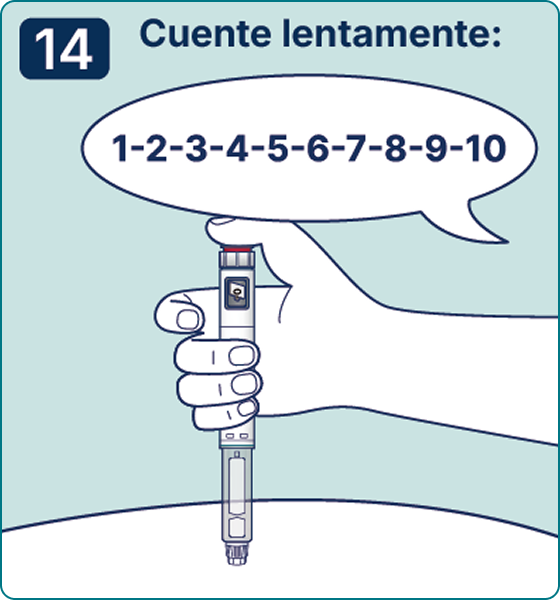
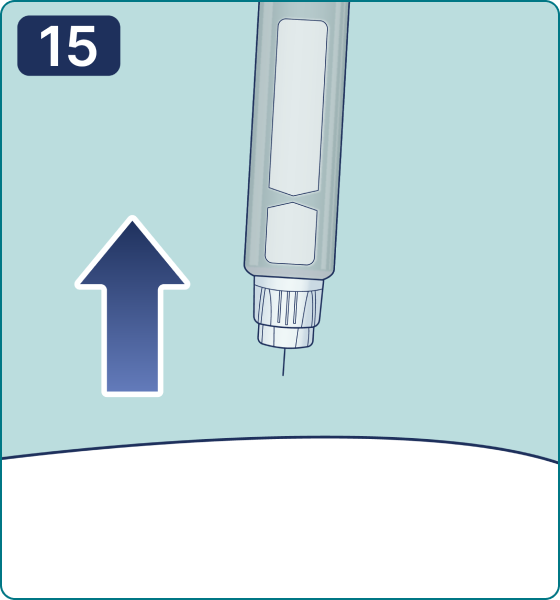
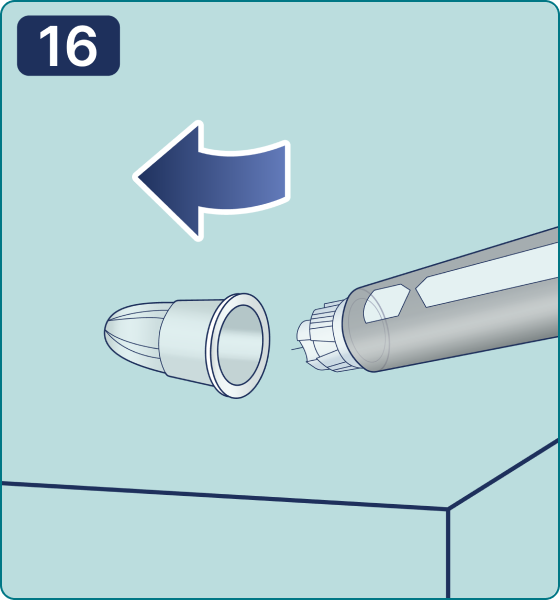
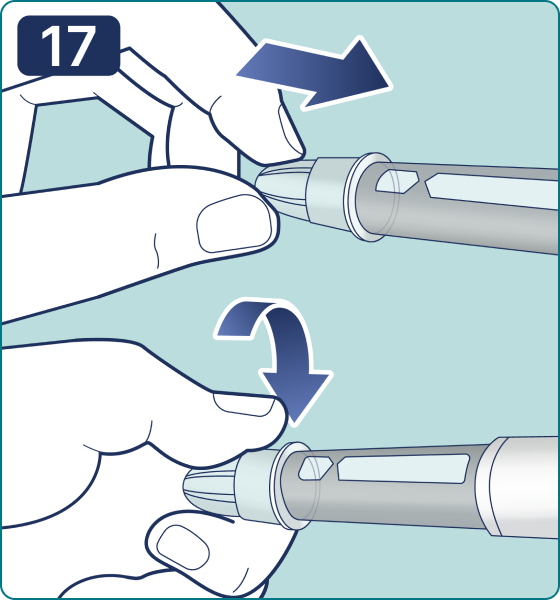
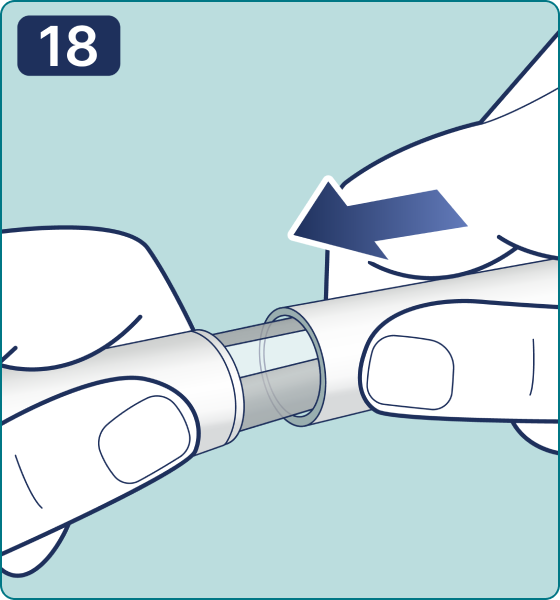
Gire el selector de dosis hasta que aparezca el símbolo « » de comprobación de flujo en el contador de dosis. Escuchará 2 (dos) clics.
» de comprobación de flujo en el contador de dosis. Escuchará 2 (dos) clics.
Nota: Si el selector de dosificación se desplazó más allá del símbolo requerido, simplemente gírelo en la dirección opuesta para corregir la posición del símbolo. Si la pluma jeringa ya está en uso,
comience a usarla desde el paso “Selección de la dosis”.